
LONG-CHAIN FATTY ACID OXIDATION DISORDERS (LC-FAOD) ARE A GROUP OF RARE, OFTEN SEVERE, LIFE-THREATENING DISORDERS THAT FEATURE UNPREDICTABLE AND PRECIPITOUS DECOMPENSATION.1,2
LC-FAOD result from defective enzymes involved in the transport and catabolism of long-chain fatty acids (LCFAs), and include the following types:3-6
CAUSE
Mutation in the CPT1A gene; prevents long-chain fatty acids from being transported into the cells’ mitochondria for breakdown
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Birth to 18 months
liver damage, low blood sugar (hypoglycemia) with low ketones (hypoketotic), seizures
CAUSE
Mutation in the SLC25A20 gene; prevents long-chain fatty acids from being transported into the cells’ mitochondria for breakdown
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Neonatal/infantile presentation
low blood sugar (hypoglycemia) with low ketones (hypoketotic), high ammonia levels in blood (hyperammonemia), enlarged liver (hepatomegaly), heart muscle damage (cardiomyopathy) with or without irregular heartbeat (arrhythmia), breathing difficulties, muscle weakness, seizures
Later onset
has been reported with milder symptoms
CAUSE
Mutation in the CPT2 gene; prevents long-chain fatty acids from being transported into the cells’ mitochondria for breakdown
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Neonatal/infantile presentation
low blood sugar (hypoglycemia) with low ketones (hypoketotic), heart muscle damage (cardiomyopathy)
Adolescent/young adult presentation
recurrent muscle breakdown (rhabdomyolysis)
CAUSE
Mutation in the ACADVL gene; prevents long-chain fatty acids from being broken down via fatty acid beta-oxidation
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Overall
heart muscle damage (cardiomyopathy) at any age
Early childhood presentation
low blood sugar (hypoglycemia) with low ketones (hypoketotic), high ammonia levels in blood (hyperammonemia)
Adolescent/adult presentation
recurrent muscle breakdown (rhabdomyolysis) and myoglobin in the urine (myoglobinuria), which causes kidney injury
CAUSE
Mutations in both the HADHA and HADHB genes, leads to defects in the entire TFP complex. Prevents long-chain fatty acids from being broken down via fatty acid beta-oxidation
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Overall
severe nerve damage (peripheral neuropathy), retina damage (retinopathy)
Early childhood presentation
similar to LCHAD deficiency but often more severe: low blood sugar (hypoglycemia) with low ketones (hypoketotic), high ammonia levels in blood (hyperammonemia)
CAUSE
Mutation in the HADHA gene, which encodes for a subunit of TFP. Prevents long-chain fatty acids from being broken down via fatty acid beta-oxidation
ESTIMATED INCIDENCE
CLINICAL PRESENTATION
Overall
skeletal muscle damage (myopathy) with or without recurrent muscle breakdown (rhabdomyolysis), nerve damage (peripheral neuropathy), retina damage (retinopathy)
Early childhood presentation
low blood sugar (hypoglycemia) with low ketones (hypoketotic), high ammonia levels in blood (hyperammonemia)
Adolescent/adult presentation
recurrent muscle breakdown (rhabdomyolysis) and myoglobin in the urine (myoglobinuria), which causes kidney injury
THE SYMPTOMS OF
LC-FAOD ARE
MULTISYSTEMIC AND
SERIOUS1,2,3,10
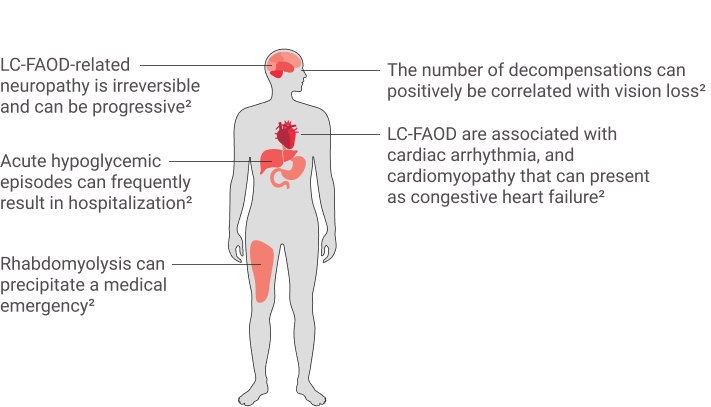
Patients CAN experience recurring metabolic crises or acute symptom episodes and can be associated with certain triggers1
Triggers1,2
- Cold temperatures
- Dehydration
- Fasting
- Hormones/menstruation
- Hot weather
- Illness/infection
- Lack of sleep
- Physiological stress
- Prolonged exercise
- Psychological stress
- Anesthetics
OCCURENCE
- 1-2 times per year
Can result in
- Clinical complications
- ER visits
- Hospitalization
Precipitating events
- Holidays/vacations
- Special events
- Any new schedule or environment in which caregivers/patients must adapt care routine or have limited access to physicians or dietitians
Living with VLCAD
Watch this video featuring Jana, a caregiver to her son Abdullah, who is living with VLCAD (Very Long-Chain Acyl-CoA Dehydrogenase deficiency)
Living with CPT II
Watch this video about a Canadian’s experience living with CPT2 (a longchain fatty acid oxidation disorder type).
References
1. Williams-Hall R, Tinsley K, Kruger E, et al. Ther Adv Endocrinol Metab.. 2022;13:1–17. 2. Merritt JL 2nd, MacLeod E, Jurecka A, Hainline B. Rev Endocr Metab Disord.. 2020;21(4):479-493. 3. Knottnerus SJG, Bleeker JC, Wüst RCI, et al. Rev Endocr Metab Disord. . 2018;19(1):93-106. 4. Wajner M, Amaral AU. Biosci Rep.. 2015;36(1):e00281. 5. Lindner M, Hoffmann GF, Matern D.J Inherit Metab Dis. .2010;33(5):521-526. 6. Wanders RJ, Ruiter JP, IJLst L, Waterham HR, Houten SM. J Inherit Metab Dis.. 2010;33(5):479-494. 7. Vockley J. Am J Manag Care. 2020;26(suppl 7):S147-S154. 8. Pennisi EM, Garibaldi M, Antonini G. J Clin Med.. 2018;7(12):E472. 9. Vitoria I, Martín-Hernández E, Peña-Quintana L, et al. JIMD Rep.. 2015;20:11-20. 10. Vockley J, Marsden D, McCracken E, et al. [published correction appears inMol Genet Metab.. 2015 Nov;116(3):221]. 11. Saudubray JM, Martin D, de Lonlay P, et al. J Inherit Metab Dis.. 1999;22(4):488-502. 12. Shekhawat PS, Matern D, Strauss AW. Pediatr Res. . 2005;57(5 Pt 2):78R-86R. 13. Vockley J, Burton B, Berry GT, et al. Mol Genet Metab.. 2017;120(4):370-377. 14. Siddiq S, Wilson BJ, Graham ID, et al. Orphanet J Rare Dis.. 2016;11(1):168.