
LES TROUBLES D’OXYDATION DES ACIDES GRAS À LONGUE CHAÎNE (LC-FAOD) SONT UN GROUPE DE TROUBLES RARES, SOUVENT GRAVES ET POTENTIELLEMENT MORTELS, QUI SE CARACTÉRISENT PAR UNE DÉCOMPENSATION IMPRÉVISIBLE ET PRÉCIPITÉE1,2.
Les LC-FAOD sont causés par des enzymes défectueuses participant au transport et au catabolisme des acides gras à longue chaîne (AGLC), et appartiennent aux types suivants3-6:
CAUSE
Mutation du gène CPT1A; empêche le transport des acides gras à longue chaîne dans les mitochondries cellulaires pour être décomposés
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Tableau clinique chez les nouveau-nés/enfants
hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), dommages au muscle cardiaque (cardiomyopathie)
Tableau clinique chez les adolescents/jeunes adultes
dégradation musculaire récidivante (rhabdomyolyse)
CAUSE
Mutation du gène SLC25A20; empêche le transport des acides gras à longue chaîne dans les mitochondries cellulaires pour être décomposés
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Tableau clinique chez les nouveau-nés/enfants
hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), taux sanguin élevé d’ammoniaque (hyperammoniémie), hypertrophie du foie (hépatomégalie), dommages au muscle cardiaque (cardiomyopathie) avec ou sans battements irréguliers (arythmie), respiration laborieuse, faiblesse musculaire, crises convulsives
Apparition tardive
rapportée avec symptômes légers
CAUSE
Mutation du gène CPT2; empêche le transport des acides gras à longue chaîne dans les mitochondries cellulaires pour être décomposés
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Tableau clinique chez les nouveau-nés/enfants
hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), dommages au muscle cardiaque (cardiomyopathie)
Tableau clinique chez les adolescents/jeunes adultes
dégradation musculaire récidivante (rhabdomyolyse)
CAUSE
Mutation du gène ACADVL; empêche la dégradation des acides gras à longue chaîne par ß-oxydation des acides gras
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Dans l’ensemble
dommages au muscle cardiaque (cardiomyopathie) à tout âge
Tableau clinique durant la petite enfance
hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), taux sanguin élevé d’ammoniaque (hyperammoniémie)
Tableau clinique chez les adolescents/adultes
dégradation musculaire récidivante (rhabdomyolyse) et myoglobine urinaire (myoglobinurie), qui cause des lésions rénales
CAUSE
Mutation des gènes HADHA et HADHB entraînant des défauts du complexe TFP complet. Empêche la dégradation des acides gras à longue chaîne par ß-oxydation des acides gras
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Dans l’ensemble
dommages nerveux graves (neuropathie périphérique), dommage à la rétine (rétinopathie)
Tableau clinique durant la petite enfance
semblable au déficit en LCHAD, mais souvent plus grave : hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), taux sanguin élevé d’ammoniaque (hyperammoniémie)
CAUSE
Mutation du gène HADHA, qui codifie la sous-unité des TFP. Empêche la dégradation des acides gras à longue chaîne par ß-oxydation des acides gras
INCIDENCE ESTIMÉE
TABLEAU CLINIQUE
Dans l’ensemble
dommages aux muscles squelettiques (myopathie) avec ou sans dégradation musculaire récidivante (rhabdomyolyse), dommages nerveux (neuropathie périphérique), dommage à la rétine (rétinopathie)
Tableau clinique durant la petite enfance
hypoglycémie (faible taux sanguin de glucose) avec faible taux de corps cétoniques (hypocétosique), taux sanguin élevé d’ammoniaque (hyperammoniémie)
Tableau clinique chez les adolescents/adultes
dégradation musculaire récidivante (rhabdomyolyse) et myoglobine urinaire (myoglobinurie), qui cause des lésions rénales
Les symptômes des LC-FAOD affectent plusieurs systèmes et sont graves1,2,3,10
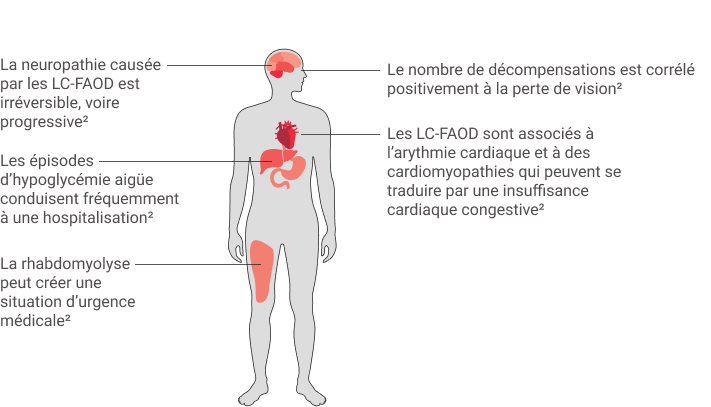
Les patients traversent des crises métaboliques ou des épisodes symptomatiques aigus récurrents, qu’ils associent à certains DÉCLENCHEURs1
DÉCLENCHEURs1,2
- Températures froides
- Déshydratation
- Jeûne
- Hormones et menstruations
- Temps chaud
- Maladie, en particulier infectieuse
- Manque de sommeil
- Stress physiologique
- Exercice prolongé
- Stress psychologique
- Anesthésiques
OCCURENCE
- 1 à 2 fois par an
CONSÉQUENCES POSSIBLES
- Complications cliniques
- Visites à l’urgence
- Hospitalisation
ÉVÉNEMENTS PRÉCIPITANTS
- Congés ou vacances
- Événements spéciaux
- Changement d’horaire ou d’environnement, quand soignants ou patients doivent modifier la routine de soins ou ont un accès restreint aux médecins ou diététiciens
Vivre avec le VLCAD
Regardez cette vidéo de Jana, qui s’occupe de son fils Abdullah, atteint de VLCAD (déficit en déshydrogénase des acyl-CoA à chaîne très longue).
Vivre avec le déficit en CPT II
Écoutez cette vidéo sur l’expérience d’une Canadienne atteinte de déficit en CPT2 (trouble de l’oxydation des acides gras à longue chaîne).
Références
1. Williams-Hall R, Tinsley K, Kruger E, et al. Ther Adv Endocrinol Metab. 2022;13:1-17. 2. Merritt JL 2nd, MacLeod E, Jurecka A, Hainline B. Rev Endocr Metab Disord. 2020;21(4):479-493. 3. Knottnerus SJG, Bleeker JC, Wüst RCI, et al. Rev Endocr Metab Disord. 2018;19(1):93-106. 4. Wajner M, Amaral AU. Biosci Rep. 2015;36(1):e00281. 5. Lindner M, Hoffmann GF, Matern D. J Inherit Metab Dis. 2010;33(5):521-526. 6. Wanders RJ, Ruiter JP, IJLst L, Waterham HR, Houten SM. J Inherit Metab Dis. 2010;33(5):479-494. 7. Vockley J. Am J Manag Care. 2020;26(suppl 7):S147-S154. 8. Pennisi EM, Garibaldi M, Antonini G. J Clin Med. 2018;7(12):E472. 9. Vitoria I, Martín-Hernández E, Peña-Quintana L, et al. JIMD Rep. 2015;20:11-20. 10. Vockley J, Marsden D, McCracken E, et al. [la correction publiée apparaît dans Mol Genet Metab., novembre 2015;116(3):221]. 11. Saudubray JM, Martin D, de Lonlay P, et al. J Inherit Metab Dis. 1999;22(4):488-502. 12. Shekhawat PS, Matern D, Strauss AW. Pediatr Res. 2005;57(5 Pt 2):78R-86R. 13. Vockley J, Burton B, Berry GT, et al. Mol Genet Metab. 2017;120(4):370-377. 14. Siddiq S, Wilson BJ, Graham ID, et al. Orphanet J Rare Dis. 2016;11(1):168.